

| Journal of Current Surgery, ISSN 1927-1298 print, 1927-1301 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Curr Surg and Elmer Press Inc |
| Journal website http://www.currentsurgery.org |
Original Article
Volume 3, Number 2, October 2013, pages 66-72
Surgical Management of Pheochromocytoma in a Contemporary Setting
Dixon T.S. Woona, b, c, Jonathan W. Serpella, Meei Yeunga, Simon Grodskia, Nathan Lawrentschuka, b, William R. Johnsona
aMonash University Endocrine Surgery Unit (MUESU), Australia
bUniversity of Melbourne Department of Surgery and Ludwig Institute Cancer Research, Austin Hospital, Australia
cCorresponding author: Dixon T.S. Woon, Western Health, 160 Gordon st. Footscray 3011, Victoria, Australia
Manuscript accepted for publication July 11, 2013
Short title: Surgical Management of Pheochromocytoma
doi: https://doi.org/10.4021/jcs188w
| Abstract | ▴Top |
Background: Pheochromocytomas are rare, with a reported incidence of 2 - 8 cases per million persons, per year. The clinical presentation of pheochromocytomas is highly variable, making diagnosis challenging. Occasionally, the potent effects of the catecholamines can also cause “pheochromocytoma crisis” which can be fatal with around 800 deaths in the United States annually. The aim of this project is to study the clinical presentation, sensitivity of biochemical tests and imaging studies, preoperative work-up, pathological findings and surgical results of patients with pheochromocytoma.
Methods: A prospective adrenalectomy database was reviewed from 1995 to 2009. The sub-group of patients with pheochromocytomas was identified. Their clinical findings, serum and urinary biochemistry results, imaging results, pre-op medical treatments, surgical approach, histopathology, and complications were reviewed.
Results: A total of 88 patients with 85 having adrenalectomy were identified. Of these 24 had pheochromocytomas with a mean age of 50 (range: 19 - 75); 14 males and ten females. Elevated urinary catecholamines (> 2 × upper normal limit) were identified in 90% of cases whilst CT imaging accurately localized all lesions (22 adrenal, 2 extra-adrenal). Pre-op medication: 75% (n = 18) required only single alpha-blockade agent, 25% (n = 6) of them combined alpha and beta-blockade. Surgery: 18 of the 22 adrenalectomies were attempted laparoscopically with 15 (83%) completed laparoscopically and the remainder (three) converted to open surgery due to adhesions or large tumor size. Four adrenalectomies were performed as planned open cases (two bilateral; two with tumors > 7.5 cm). All patients had benign pheochromocytomas. One patient developed post-operative pneumonia and one patient had post-op hypotension needing inotropic support for two days. All patients are alive and well, with no evidence of recurrent disease (minimum two year follow-up).
Conclusions: The majority of adrenalectomies may be safely performed laparoscopically with minimal complications. A multi-disciplinary approach contributes to successful outcomes with involvement extending beyond preoperative consideration of the diagnosis into accurate localization of lesions, medical treatment when appropriate, specialized anaesthetic involvement and intensive care support.
Keywords: Pheochromocytoma; Hypertension; Laparoscopic surgery; Adrenalectomy; Adrenal neoplasm
| Introduction | ▴Top |
Pheochromocytomas are neuroendocrine tumors derived from chromaffin cells of the adrenal medulla. Extra-adrenal pheochromocytomas are called paragangliomas and they account for 15 - 20% of all pheochromocytoms [1]. Pheochromocytomas are rare, with a reported incidence of 2 - 8 cases per million persons, per year [2]. Patients with sporadic pheochromocytomas are usually diagnosed at age 40 - 50 years [1, 3]. Patients with familial cancer syndromes such as multiple endocrine neoplasia (MEN) type 2, von Hippel-Lindau disease, and neurofibromatosis type 1 (von Recklinghausen’s disease) are at much higher risk of developing pheochromocytomas [4]. These patients are usually diagnosed with pheochromocytomas at age 30 - 40 years, and they are more likely to have multifocal disease [5].
Pheochromocytoma has been termed the ‘10% tumor’; with 10% bilateral, 10% malignant, 10% extra-adrenal, 10% hereditary, and 10% paediatric [6]. These tumors produce catecholamines, usually adrenaline, noradrenaline or dopamine. The uncontrolled secretion of these catecholamines is often symptomatic where such patients, may experience more commonly headache, palpitations, sweating and less often nausea, flushing, weight loss, tiredness, anxiety and panic attacks [4]. It is estimated that pheochromocytomas occur in 0.1-0.5% of patients presenting with hypertension [1, 4, 7]. However, 15% of patients are asymptomatic and the diagnosis is made incidentally on imaging investigating other conditions [8].
Rarely, the potent effects of the catecholamines can also cause “pheochromocytoma crisis” which can be fatal with around 800 deaths in the United States annually [1]. The crisis typically manifests as severe hypertention, cardiogenic shock, acute myocardial infarction, pulmonary oedema, encephalopathy and multiorgan failure [9, 10]. Precipitants include needle biopsies, angiography (intravenous contrast), general anaesthesia, and surgery. About a third of sudden deaths, secondary to pheochromocytomas, occur during or after an operative procedure or delivery [6].
Biochemical tests to diagnose pheochromocytomas include the measurement of urinary or plasma catecholamines. Measurement of the 24-hour urinary excretion of catecholamines (and its metabolites) has a sensitivity of 86% and measurement of plasma catecholamines has a sensitivity of 84%[11] Imaging techniques such as CT or MRI and functional studies such as (123)I- Metaiodobenzylguanidine (MIBG) scintigraphy are used to localise the tumors [1].
Complete surgical resection of pheochromocytoma is usually curative. In the past, surgical mortality associated with excision of pheochromocytomas was as high as 50% [12, 13]. More recent reviews report mortality rates to be less than 3% [1]. Improved surgical outcomes have been the result of advances in localization techniques, perioperative medical management and anaesthetic practice [13]. We report our recent experience in the management of pheochromocytoma to highlight that surgical management of pheochromocytoma can be performed safely with modern perioperative management.
| Materials and Methods | ▴Top |
From 1995 to 2009, eighty-eight patients were referred to the Monash University Endocrine Surgery Unit (MUESU) for adrenalectomy. Patient demographics, clinical presentation, results of serum or urine biochemistry, results of imaging, preoperative medical treatments, surgery performed, histopathology, morbidity and mortality were recorded in the prospective adrenalectomy database with ethical approval. The data was then analysed with a focus on identifying patients having a pheochromocytoma. Their clinical presentation, methods of confirming the diagnosis of pheochromocytoma, imaging findings, details of pre-operative work-up, pathology findings, surgical approach, and surgical outcomes were reviewed.
| Results | ▴Top |
Patient population
There were 88 patients in the adrenalectomy database from 1995 to 2009. Of these, 85 patients had adrenalectomies performed. Of the three patients who did not have adrenalectomy: one had a gastric diverticulum mimicking a left adrenal mass discovered during operation and adrenalectomy was not performed; one was found to have metastatic disease (primary lung cancer) in liver during operation; and one patient had bilateral metastatic adrenal carcinomas. The histological diagnosis of the 85 patients who had adrenalectomies is detailed in Table 1. Benign cortical adenoma was the most common histological diagnosis with 37 cases in total. In this group, 21 cases presented as incidentalomas (incidental findings on imaging); 13 cases presented as Conn’s syndrome and three cases presented as Cushing’s syndrome. The second most common histological diagnosis was pheochromocytoma. There were 24 (28%) patients with pheochromocytomas. The mean age of patients with pheochromocytomas was 50 (range: 19 - 75), 33% (8/24) of patients were under the age of 50 and 42% (10/24) were in their fifties. There were 14 male and 10 female patients. Ten of the pheochromocytomas were left-sided, 12 were right-sided and two were bilateral.
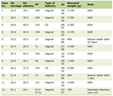 Click to view | Table 1. Histology Findings of 85 Adrenalectomies |
Clinical presentation
As patients with pheochromocytomas have varying presentations, they were categorised into three groups according to their clinical presentations (Table 2). The “investigation for hypertension” group consisted of patients with hypertension who were investigated for a secondary cause for hypertension. There were 13 (43%) patients in this group. Of these patients with hypertension, eight were symptomatic. Their primary symptoms are shown in Table 2. Only two patients had the combination symptoms of flushing, palpitation and anxiety. Five patients in this group were asymptomatic. The “incidentaloma” group consisted of patients diagnosed with adrenal masses during imaging for an unrelated clinical presentation with no previous symptoms related to pheochromocytoma. There were nine (37.5%) patients in this group. The “screening” group consisted of patients with a known family history of pheochromocytoma, or known familial syndrome associated with an increased risk of pheochromocytoma developing. Two patients were in this group. One patient has Von Recklinghausen’s disease and one has MEN 2A syndrome.
Click to view | Table 2. Clinical Presentations of Patients With Pheochromocytomas |
Tumor biochemistry
All 24 patients had 24-hour urinary collection for catecholamines (adrenaline, noradrenaline or dopamine). A catecholamine level of more than twice the upper limit of normal was considered diagnostic. If the first set of urinary catecholamines result was normal and the diagnosis of pheochromocotoma was still suspected, a 24-hour urinary collection for catecholamines was repeated. Only 21 patients had complete urinary catechlamines data for analysis. Two patients’ results were not found. One patient had elevated urinary catecholamines but the exact number was not found (Table 3). Based on the 21 results, 19 (90%) patients had greater than 2 × upper limit of normal catecholamines levels (adrenaline, noradrenaline or dopamine) in their urine. Therefore, the sensitivity of detecting pheochromocytomas was 90%. Two patients had urinary catecolamine levels only 1.5 ×the upper limit of normal. One of these two patients has Von Recklinghausan syndrome. The other patient had a long history of hypertension. MIBG scintigraphy confirmed the diagnosis of pheochromocytoma for both of these patients.
Click to view | Table 3. Urinary Catecholamines Levels in Patients With Pheochromocytomas |
Imaging
All patients (n = 24) had CT scans, which correctly localised tumors (22 adrenal, 2 extra-adrenal). Measurement of the adrenal tumors consisted of length, width, and height. The mean maximal CT measured size was 4.3 cm (range from 2 cm to 9 cm).
A total of 18 patients had a MIBG scintigraphy. MIBG scintigraphy correctly detected 14 unilateral adrenal pheochromocytomas, one bilateral adrenal pheochromocytoa, and two extra-adrenal pheochromocytomas. MIBG scintigraphy failed to detect only one (5.6%) pheochromocytoma (this patient had a 2.5 cm left adrenal mass on CT and an elevated urinary catecholamine).
In general, MRI scan was requested for identifying possible vascular invasion or thrombus formation. Seven patients had MRI scans, all correctly localized the lesions. The MRI scans correctly confirmed that none of these patients had vascular invasion or evidence of thrombus.
Pre-operative medical management: All patients were referred to the endocrinology and anaesthesia services before surgery. When indicated, patients were seen by cardiology. Before surgery, all patients were given an alpha-blockading agent. The dose of alpha-blockage agent was titrated until postural hypotension was achieved. A beta-blockade agent was added if the patient displayed tachycardia. In total, 75% of patients’ blood pressure was successfully managed with only alpha-blockage (phenoxybenzamine for 14 patients and prozosin for four patients) whilst 25% of patients required both alpha and beta-blocking agent (Five patients required phenoxybenzamine and a beta blocker and one patient was prescribed prazosin and propanolol). Surgical approach: 22 patients had adrenal pheochromocytomas and two patients had extra-adrenal pheochromocytomas. Patients with adrenal pheochromocytomas had either open or laparoscopic adrenalectomies (Table 4), 18 of these 22 were planned to have laparoscopic adrenalectomies, 15 of these 18 (83%) adrenalectomies were successfully completed laparoscopically. Three (16.7%) laparoscopic adrenalectomies were converted to open. One was due to a very large tumor of 10 cm in size. One was due to extensive adhesions and fat necrosis. One was due to difficult anatomy and dissection. The four patients with adrenal pheochromocytomas who were not suitable for laparoscopic approach had open adrenalectomies. Two were bilateral tumors and two were large tumors (9 cm and 7.6 cm respectively). The two patients with extra-adrenal pheochromocytomas had open resection of pheochromocytomas.
Click to view | Table 4. Operative Approach |
Histopathology
Mean tumor size of these pheochromocytomas was 5.7 cm (range 20 cm to 15 cm). All were benign pheochromocytomas and all had clear margins of resection.
Complications
One patient (4%) developed post-op pneumonia and another (4%) post-op hypotension requiring inotropic support for two days. However, all patients are still alive and well, without evidence of recurrent disease on follow-up.
| Discussion | ▴Top |
Pheochromocytoma is uncommon with a highly variable clinical presentation making diagnosis challenging. In this series, classical symptoms such as headache, flushing, sweating, palpitations or anxiety attacks only occurred in one third of the patients; and only half of the patients had hypertension. Incidentalomas accounted for 38% of pheochromocytomas in this study. A review of the coronial autopsy records from Melbourne, Sydney and Auckland had shown that a significant number of pheochromocytomas were not diagnosed during life, and this may have contributed to the patient’s death in up to 50% of the cases [14]. Furthermore, anaesthesia and surgery in the presence of undiagnosed pheochromocytoma might be the cause of death in up to 25% of cases [15].
However, once the diagnosis of pheochromocytoma is confirmed, elective resection is safe. Therefore, it is important to have a high index of suspicion, and offer biochemical tests to all patients with suspected pheochromocytoma. These include patients with intermittent signs or symptoms suggestive of a pheochromocytoma; patients with treatment resistant hypertension; patients with cardiovascular instability during anaesthesia and surgery; patients with genetic predispositions to pheochromocytoma such as MEN type 2, von Hippel Lindau syndrome, neurofibromatosis type 1 and mutations of the succinate dehydrogenase genes; all patients with incidentalomas; and patients with sudden attacks of anxiety [1]. Unusual presentations of pheochromocytoma such as myocardial infaction, cardiomyopathy, cardiogenic shock, pulmonary oedema, pulmonary hemorrahage, cyclic hypotension, encephalopathy and renal failure had also been reported [10, 16]. Therefore, the diagnosis of pheochromocytoma must be considered when the underlying etiology of patients with these presentations is unclear.
The most commonly used biochemical testing in our centre to diagnose pheochromocytomas is 24-hour urinary collection for catecholamines (adrenaline, noradrenaline or dopamine). This study showed that the sensitivity of urinary catecholamines was 90%. Two patients’ (9.5%) 24-hour urinary cathecholamine levels were 1.5 × the upper limit of normal. One of these patients had Von Recklinghausan syndrome with a tumor of 2 cm and one had a long history of hypertension with a tumor of 3 cm on CT scan. Due to clinical suspicion of pheochromocytoma despite sub-diagnostic urinary catecholamines level, both patients had MIBG scintigraphy and the diagnosis of pheochromocytoma was confirmed. It is well documented that the sensitivity value of biochemical tests for hereditary pheochromocytoma is lower [11]. This is because routine screening for pheochromocytoma in patients with a hereditary predisposition to the tumor often leads to detection of small tumors that release lower level of catecholamines. Other studies had suggested the use of plasma-free metanephrines (99% sensitivity) or urinary-fractionated metanephrines (97% sensitivity) as the test of first choice for diagnosing pheochromocytoma [1, 11]. CT scanning and MRI have similar sensitivities (> 90%) and specificities (79-80%) [1, 17, 18]. However, CT scan is still the method most commonly used to evaluate patients suspected of having adrenal pathology [19, 20], and it is the preferred initial imaging modality in diagnosing pheochromocytoma in our centre. It correctly localised all pheochromocytomas in this series. However, CT is less helpful for extra-adrenal lesions and recurrent tumors [21]. When these lesions are suspected, MIBG scintigraphy is usually performed. MIBG scintigraphy plays an important role in identifying extra-adrenal, additional multifocal or metastatic turmors, and recurrent tumors. It is most relevant in patients with extra-adrenal or large (> 5 cm) adrenal tumor with increase risk of malignant disease or in patients with high suspicion of the presence of multifocal disease such as those with a familial syndrome [1, 22].
MRI can show more clearly than CT the relations of adrenal masses to adjacent vascular structures. The sensitivity is reported to be up to 100% with a positive and negative predictive value of 83% and 100% respectively [21]. Therefore, MRI is used to identify possible vascular invasion or thrombus formation.
Before the elective resection of pheochromocytomas, all patients were seen by our endocrinologists and anaesthetists. Patients were also referred to cardiology if they had a history of ischaemic heart disease, cardiomyopathy or any other significant cardiac conditions. These pre-operative medical management and the support from intensive care unit were some of the reasons that pheochromocytoma operations were carried out safely with a low complication rate in our centre.
In the past decade laparoscopic adrenalectomy has become the preferred surgical approach for adrenalectomy, even for pheochromocytoma. Recent studies had confirmed that laparoscopic adrenalectomy is a safe and effective treatment for pheochromocytoma [23-28]. Patients who had laparoscopic adrenalectomy for pheochromocytomas had shorter mean operating time and hospital stay; less intraoperative hypertension or intraoperative blood loss; less postoperative analgesia requirement, and earlier return to oral nutrition [26]. Up to 90% of suitable cases (adrenal pheochromocytoma with no suspicion of malignancy and a reasonable size tumor) could be completed laparoscopically [29]. In this study, 83% of our planned laparoscopic adrenalectomy was completed laparoscopically and with minimal complication.
In conclusion, surgery for patients with pheochromocytoma can be safely carried out with minimal complications. This was achieved with careful consideration of the pre-operative diagnosis, localization of lesions with imaging, preoperative medical treatment, and a multidisciplinary approach utilising endocrinologists, cardiologists, intensive care physicians and specialized anesthetists.
Grant Support
Nil.
Financial Disclosures
Nil.
| References | ▴Top |
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665-675.
doi - Harding JL, Yeh MW, Robinson BG, Delbridge LW, Sidhu SB. Potential pitfalls in the diagnosis of phaeochromocytoma. Med J Aust. 2005;182(12):637-640.
pubmed - Goldstein RE, O'Neill JA, Jr., Holcomb GW, 3rd, Morgan WM, 3rd, Neblett WW, 3rd, Oates JA, Brown N, et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg. 1999;229(6):755-764; discussion 764-756.
- Petri BJ, van Eijck CH, de Herder WW, Wagner A, de Krijger RR. Phaeochromocytomas and sympathetic paragangliomas. Br J Surg. 2009;96(12):1381-1392.
doi pubmed - Neumann HP, Berger DP, Sigmund G, Blum U, Schmidt D, Parmer RJ, Volk B, et al. Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease. N Engl J Med. 1993;329(21):1531-1538.
doi pubmed - Thompson G. Cameron: Current Surgical Therapy, 9th ed. 2008.
- Williams DT, Dann S, Wheeler MH. Phaeochromocytoma—views on current management. Eur J Surg Oncol. 2003;29(6):483-490.
doi - Amar L, Servais A, Gimenez-Roqueplo AP, Zinzindohoue F, Chatellier G, Plouin PF. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J Clin Endocrinol Metab. 2005;90(4):2110-2116.
doi pubmed - James MF, Cronje L. Pheochromocytoma crisis: the use of magnesium sulfate. Anesth Analg. 2004;99(3):680-686, table of contents.
doi pubmed - Kizer JR, Koniaris LS, Edelman JD, St John Sutton MG. Pheochromocytoma crisis, cardiomyopathy, and hemodynamic collapse. Chest. 2000;118(4):1221-1223.
doi pubmed - Lenders JW, Pacak K, Walther MM, Linehan WM, Mannelli M, Friberg P, Keiser HR, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA. 2002;287(11):1427-1434.
doi pubmed - Manger WM, Gifford RW, Jr., Hoffman BB. Pheochromocytoma: a clinical and experimental overview. Curr Probl Cancer. 1985;9(5):1-89.
pubmed - Ulchaker JC, Goldfarb DA, Bravo EL, Novick AC. Successful outcomes in pheochromocytoma surgery in the modern era. J Urol. 1999;161(3):764-767.
doi - McNeil AR, Blok BH, Koelmeyer TD, Burke MP, Hilton JM. Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne and Auckland. Aust N Z J Med. 2000;30(6):648-652.
doi pubmed - Platts JK, Drew PJ, Harvey JN. Death from phaeochromocytoma: lessons from a post-mortem survey. J R Coll Physicians Lond. 1995;29(4):299-306.
pubmed - Park M, Hryniewicz K, Setaro JF. Pheochromocytoma presenting with myocardial infarction, cardiomyopathy, renal failure, pulmonary hemorrhage, and cyclic hypotension: case report and review of unusual presentations of pheochromocytoma. J Clin Hypertens (Greenwich). 2009;11(2):74-80.
doi pubmed - Ilias I, Pacak K. Current approaches and recommended algorithm for the diagnostic localization of pheochromocytoma. J Clin Endocrinol Metab. 2004;89(2):479-491.
doi - Lumachi F, Tregnaghi A, Zucchetta P, Cristina Marzola M, Cecchin D, Grassetto G, Bui F. Sensitivity and positive predictive value of CT, MRI and 123I-MIBG scintigraphy in localizing pheochromocytomas: a prospective study. Nucl Med Commun. 2006;27(7):583-587.
doi pubmed - Falke TH, te Strake L, Shaff MI, Sandler MP, Kulkarni MV, Partain CL, Nieuwenhuizen-Kruseman AC, et al. MR imaging of the adrenals: correlation with computed tomography. J Comput Assist Tomogr. 1986;10(2):242-253.
doi pubmed - Schultz CL, Haaga JR, Fletcher BD, Alfidi RJ, Schultz MA. Magnetic resonance imaging of the adrenal glands: a comparison with computed tomography. AJR Am J Roentgenol. 1984;143(6):1235-1240.
doi pubmed - Safwat AS, Bissada NK, Seyam RM, Al Sobhi S, Hanash KA. The clinical spectrum of phaeochromocytoma: analysis of 115 patients. BJU Int. 2008;101(12):1561-1564.
doi pubmed - Milardovic R, Corssmit EP, Stokkel M. Value of 123I-MIBG Scintigraphy in Paraganglioma. Neuroendocrinology. 2010;91(1):94-100.
doi pubmed - Perry KA, El Youssef R, Pham TH, Sheppard BC. Laparoscopic adrenalectomy for large unilateral pheochromocytoma: experience in a large academic medical center. Surg Endosc. 2010;24(6):1462-1467.
doi pubmed - Castilho LN, Simoes FA, Santos AM, Rodrigues TM, dos Santos Junior CA. Pheochromocytoma: a long-term follow-up of 24 patients undergoing laparoscopic adrenalectomy. Int Braz J Urol. 2009;35(1):24-31; discussion 32-25.
- Tiberio GA, Baiocchi GL, Arru L, Agabiti Rosei C, De Ponti S, Matheis A, Rizzoni D, et al. Prospective randomized comparison of laparoscopic versus open adrenalectomy for sporadic pheochromocytoma. Surg Endosc. 2008;22(6):1435-1439.
doi pubmed - Toniato A, Boschin IM, Opocher G, Guolo A, Pelizzo M, Mantero F. Is the laparoscopic adrenalectomy for pheochromocytoma the best treatment? Surgery. 2007;141(6):723-727.
doi pubmed - Thomson BN, Moulton CA, Davies M, Banting SW. Laparoscopic adrenalectomy for phaeochromocytoma: with caution. ANZ J Surg. 2004;74(6):429-433.
doi pubmed - Meyer-Rochow GY, Soon PS, Delbridge LW, Sywak MS, Bambach CP, Clifton-Bligh RJ, Robinson BG, et al. Outcomes of minimally invasive surgery for phaeochromocytoma. ANZ J Surg. 2009;79(5):367-370.
doi pubmed - Solorzano CC, Lew JI, Wilhelm SM, Sumner W, Huang W, Wu W, Montano R, et al. Outcomes of pheochromocytoma management in the laparoscopic era. Ann Surg Oncol. 2007;14(10):3004-3010.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Current Surgery is published by Elmer Press Inc.